
Simon Lande
Rare Disease Day is fast approaching, with a programme of activity being lined up, through international initiatives and country-specific programmes, in the UK for example to highlight challenges, raise awareness and generate change for the 300 million people worldwide living with one or more of the roughly 8,000 known rare diseases, as well as their families and carers.
A substantial portion of people with rare diseases face long delays in getting an accurate diagnosis; symptoms may be non-specific, overlap with more common conditions, or vary significantly between patients. This leads to a high rate of misdiagnosis or delayed diagnosis, resulting in a large undiagnosed population, meaning that the total population living with such conditions may be much greater than current estimates, as illustrated in the following graphic:
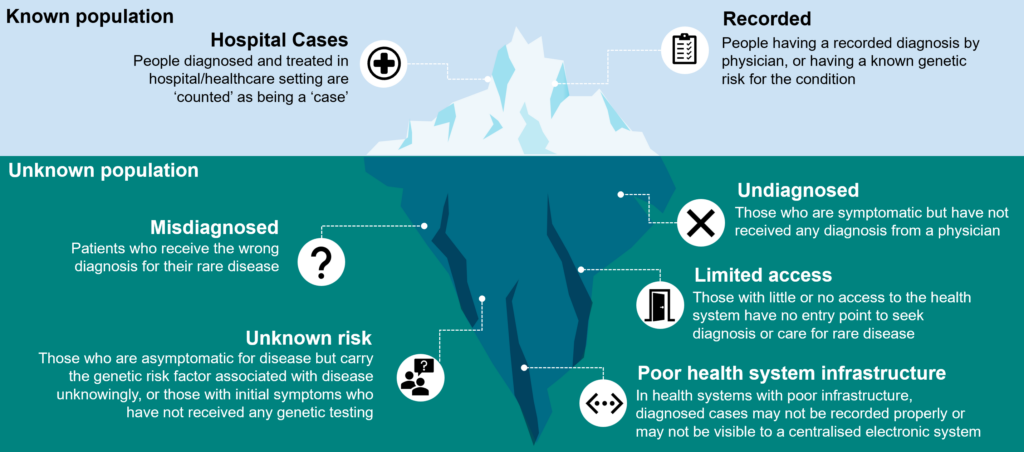
As medical knowledge advances, new rare diseases are identified, and existing diseases are better understood, which can change the number of undiagnosed individuals.
There is also no universally agreed definition of what constitutes a rare disease. Different countries have different thresholds for considering a disease rare.
Furthermore, as there are thousands of rare diseases, many of them are poorly understood, and this diversity makes it difficult to gather comprehensive data. Obtaining accurate data is further made more challenging due to the lack of reporting and data collection: many countries do not have specific registries or reporting systems for rare diseases, making it hard to track cases accurately.
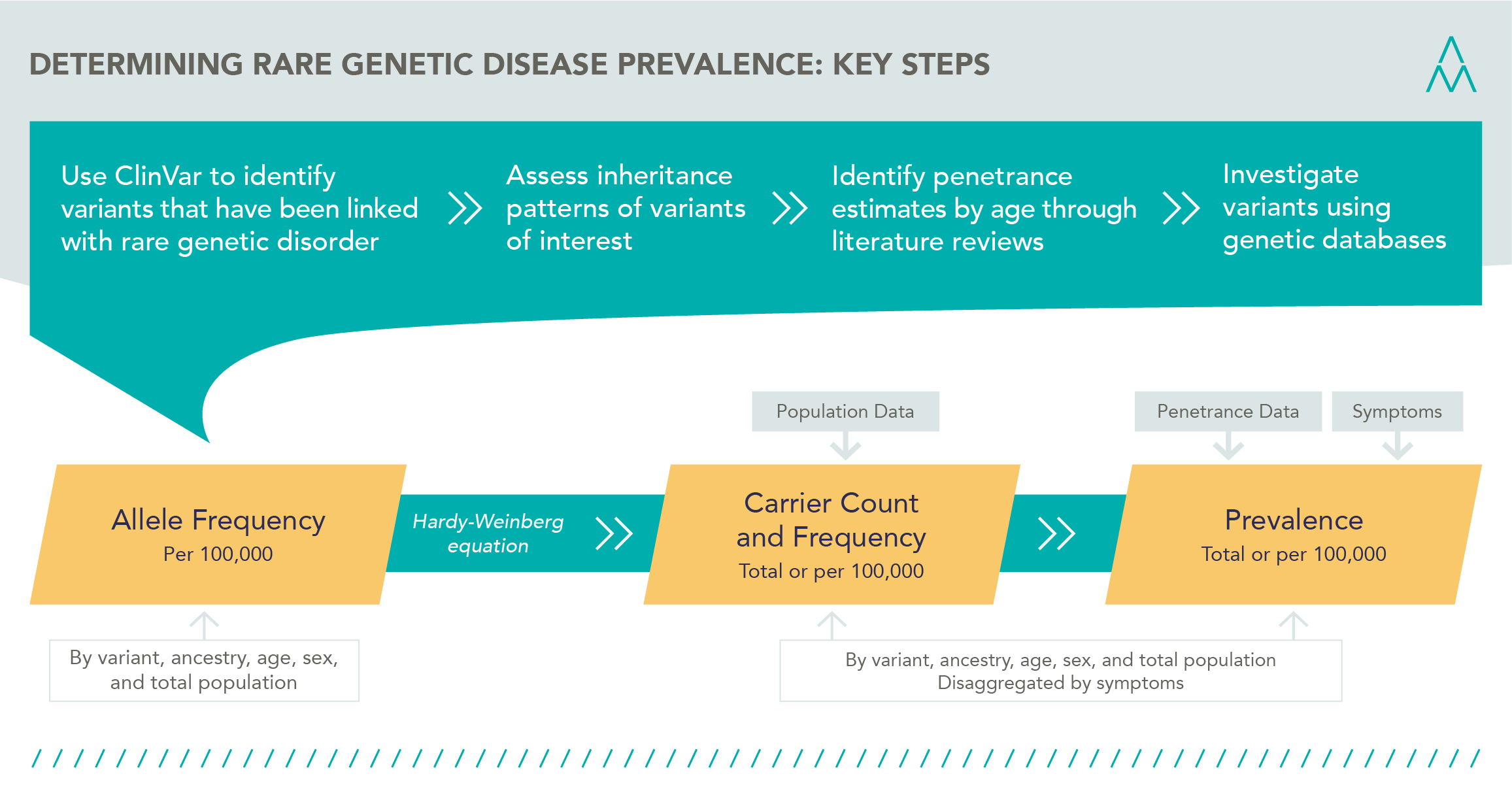
As covered in some previous blog posts, at HealthLumen we have been working with large genetic datasets such as gnomAD and TOPMed, which enable the frequency of occurrence of a given allele to be quantified and, coupled with penetrance, this can give a more accurate estimate of the true burden of the disease, by estimating both diagnosed and undiagnosed cases. Exciting developments continue to proceed at pace in this area. For example:
Using the data from these and other databases, our methodology to determine prevalence is illustrated in the graphic below, and includes:
Senior Evidence Lead Joshua Card-Gowers will be presenting the results of some of our research at the World Orphan Drug Congress in Boston.